BIOTECH-7005-BIOINF-3000
Evolutionary Processes assignment (Due 05/09/2025)
In today’s practical we are trying to wean you off of detailed instructions and will expect you to be able to do basic things that have been covered before, such as renaming a file or using wget. You should read through the practical fully before beginning so that you understand the sequence of operations and what is expected.
Setup the directory for today
Just as we’ve created a new R Project for practicals 1 to 3, let’s create a new one for today to make sure we’re all in the same place.
- Using the
Filemenu at the top left, selectNew Project - Select
New Directory - Select
New Project - If you’re not already asked to create this project as a subdirectory of
~, navigate to the Home directory using the Browse button - In the
Directory Namespace, enterPractical_5, then hit the Create Project button.
This again helps us keep our code organised and is good practice.
Prepare the sequence data
Get the sequences from the Bioinf 3000 repository (https://university-of-adelaide-bx-masters.github.io/BIOTECH-7005-BIOINF-3000/Practicals/evolutionary_prac/bovidae_73_mtDNA.fa) using wget.
Reduce the size of the dataset
Run the following command using your student number for id:
./subset -id aXXXXXXX -n 50 -in bovidae_73_mtDNA.fa > bovidae_50_mtDNA.fa
(The subset command can be obtained from here (https://university-of-adelaide-bx-masters.github.io/BIOTECH-7005-BIOINF-3000/Practicals/evolutionary_prac/subset)
using wget.)
Remember to make subset executable (see the bash practical for a reminder on how to do this)
This selects 50 sequences from the input alignment according to your student number and writes them to a new file. This is not normal practice, and is only necessary because of prac time limits.
Software used for this practical
You will use the following software today: MAFFT for making multiple alignments read about it here, Mr Bayes for building Bayesian phylogenies read about it here, seqmagick a comprehensive toolkit for sequence conversion/manipulation read about it here and Gblocks a tool to remove poorly aligned regions from multiple alignments to improve the inference of phylogenies read about it here.
You will need to activate the bioinf conda environment so that you can use Gblocks.
source activate bioinf
Multiple alignment
In order to construct a phylogenetic tree, we need to provide positional information to the tree reconstruction program so that related positions are comparable. This is done by constructing a multiple sequence alignment. There are many programs that can be used to do this; two fast programs are MUSCLE and MAFFT, today we have installed and will be using MAFFT.
Run the following commands:
mafft bovidae_50_mtDNA.fa > bovidae_50_mtDNA-mafft.fa
Remove non-conserved blocks
Many programs unfortunately have name length limitations. One of the programs we will be using (Gblocks) cannot have long sequence identifiers and is not open source, so we cannot fix it. To get around this problem we will shorten the identifiers in a meaningful way.
Run the following command to change the sequence IDs:
sed -e 's/^>[^ ]\+ \([^ ]\+\) \([^ ]\+\).*$/>\1_\2/g' bovidae_50_mtDNA-mafft.fa > bovidae_50_mtDNA-named.fa
Q1. What does this command do?
In order to be able to examine the alignments more effectively we will convert the format from FASTA to NEXUS format. This will make it easier to see the alignment.
seqmagick convert --output-format nexus --alphabet dna bovidae_50_mtDNA-named.fa bovidae_50_mtDNA-named.nex
Look at the NEXUS alignment file. At the beginning of the alignment and near the end there are regions that have large gaps and very poor conservation. Because the mitochondrial genome is circular, the chosen “position 0” is conventionally set to the beginning of the non-coding Control Loop, which can be highly variable between species.
The phylogenetic reconstruction methods we will be using cannot handle missing bases, so these must be removed.
We can use Gblocks to remove the non-conserved regions of the alignment.
Run Gblocks by entering Gblocks.
You should see something like this:
******************************************************
GBLOCKS 0.91b
SELECTION OF CONSERVED BLOCKS FROM MULTIPLE ALIGNMENTS
FOR THEIR USE IN PHYLOGENETIC ANALYSIS
******************************************************
o. Open File
s. Saving Options
g. (Get Blocks)
b. Block Parameters
s. Saving Options
g. (Get Blocks)
q. Quit
Use the menu options in the program to remove the non-conserved regions from the fa file (not the nex file).
This will give you a file bovidae_50_mtDNA-named.fa-gb.
Q2. Why do we need to do this? (Hint:read the Gblocks documentation)
Convert to NEXUS format
The program we will be using to perform phylogenetic reconstruction uses a sequence (and other character) format called NEXUS. The NEXUS format is fairly widely used for phylogenetic data as it can be used to encode a variety characters, not limited to sequence data.
Use seqmagick to convert the fa-gb file containing the trimmed alignment blocks to a nexus format with the name bovidae_50_mtDNA-named.nex.
Note that you will need to rename the fa-gb file so that it has an fa file extension, remember not to overwrite your original bovidae_50_mtDNA-named.fa when doing so.
Look at your .nex file to satisfy yourself that things are as they should be.
Run Mr Bayes
Mr Bayes is a program for performing phylogenetic tree reconstruction from multiple sequence alignments using a Bayesean inference. The Bayesean approach to reconstruction is based on using a likelihood function to estimate posterior probabilities for trees and model parameters. There are concerns in the literature about the validity of Bayesean reconstruction, but Mr Bayes provides a good environment to do the practical and Baysean inference works for many data sets.
A manual for Mr Bayes is available from here.
Start Mr Bayes by entering the following command into a terminal:
mb
This will give you a Mr Bayes prompt that should look like this.
MrBayes v3.2.6 x64
(Bayesian Analysis of Phylogeny)
Distributed under the GNU General Public License
Type "help" or "help <command>" for information
on the commands that are available.
Type "about" for authorship and general
information about the program.
MrBayes >
If you enter help at the prompt you will get a list of topics that can be read, for example help execute.
Load sequence data
First you will need to load the data set that you prepared.
Use the execute command to load the NEXUS sequence file you created with seqmagick.
Q3. How many taxa were read into memory?
Q4. How many characters are being used?
Adjust likelihood settings
For the practical you are to perform 2 reconstructions, choosing from substitution models with 1, 2, 6 or mixed. Each run takes about an hour, so you will have time to work on other parts of the practical while it is running.
First you need to set the number of substitution rate parameters used in the model. Before you do this, see what the values of parameters are by default in the likelihood setting.
help lset
You can see that the default rate matrix is 4 by 4 which corresponds to nucleotide.
Q5. What are the models used by the default substitution type?
Set the number of substitution types to the number you have chosen (written below as <num>).
lset nst=<num>
Run Monte-Carlo Markov Chain analysis
The next thing to do is start the tree reconstruction analysis.
This is done using the mcmc (Markov chain Monte Carlo) command.
Before you do this, you will need to set some options.
First examine the defaults.
help mcmc
You can see here that the default analysis runs for 1,000,000 iterations, samping every 500th iteration for tree reconstruction. You can also see that the first 25% of samples are discarded by default.
We don’t have enough time to run for 1x10^6 generations, we want to see the convergence behaviour and we want to specify the output file name (again <num> is the model subtitution type number), so start the analysis with the following command.
mcmc ngen=50000 relburnin=no burnin=0 filename=model-<num>
This will take 5-6 minutes (the remaining time is shown to the right of the output). You will see in the output the log likelihoods of the trees being considered (2 runs by 4 chains) in the analysis, and every 5 lines the variance between the two runs is output to give an indication of how far the trees have converged. You can see that as the run continues the average standard deviation of split frequencies decreases, showing that the two runs are converging on the same tree or similar trees.
Examine convergence
After the MCMC has completed the 50,000 iterations, it will output the final average standard deviation of split frequencies and ask if the run is to be continued. Answer “no”.
See the Mr Bayes manual for a more detailed explanation of how to determine the answer to this question. I strongly suggest putting your Ctrl+F skills to work (a vital skill in bioinformatics!).
When you stop the runs, Mr Bayes will output summary data from the analysis.
To look at the convergence, execute the following command.
sump
Q6. Describe what the initial output graph shows.
Find where the plateau starts by choosing different burnin lengths (note that the burnin describes the number of sampled trees to discard, not the number of MCMC iterations). You will not be able to answer the following question without playing around with a couple of burnin values.
Q7. Approximately when does the plateau start in terms of number of samples? Provide evidence to support your answer.
Examine trees
When you are happy with the burnin - that the log likelihood has plateaued - you can get the consensus tree to be calculated.
sumt
This will output a large quantity of data about the estimated paramaters and the statistical support for branch nodes, and two trees, one showing the consensus branch lengths and one showing the branch node support. It will also output a consensus tree file called “model-<num>.con.tre”.
Repeat the analysis with another model and compare the trees using the FigTree program. FigTree is a tree visualising tool that you will need to run on your computer. Select the best download type for your computer/operating system and install it (use .dmg for Mac, use .zip for Win).
Q8. Do the trees from the two models differ?
Q9. Do the trees agree with the known taxonomic groupings? If not, propose possible explanations.
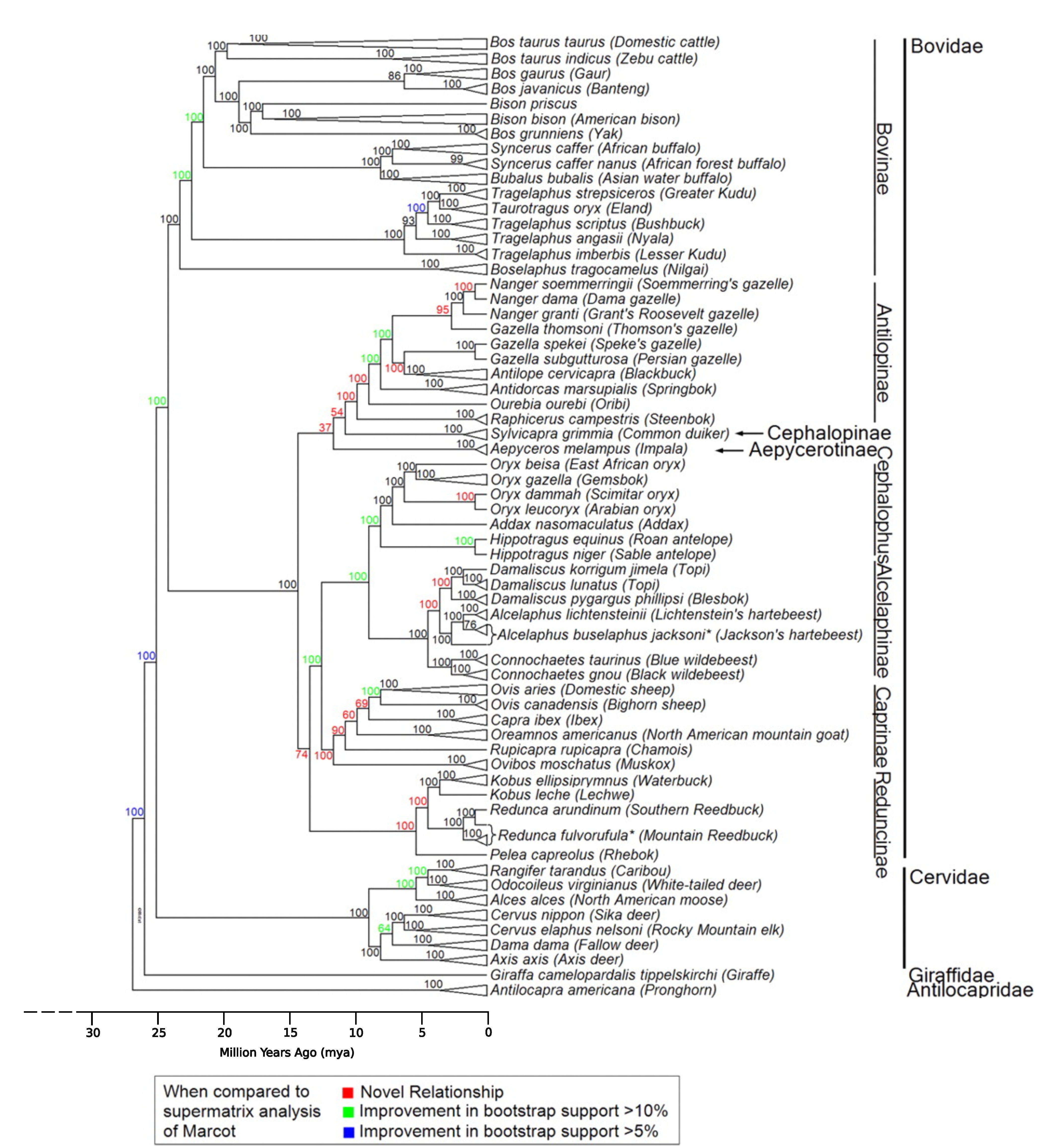
Nuclear tree from Decker et al. 2009 doi:10.1073/pnas.0904691106. A timescale has been added to show the most recent common ancestor of Ruminants existed ~27 million years ago, based on current data from TimeTree
Tasks (due 05/09/2025)
Practical questions
Answer the questions in bold above.
Bayesean Trees with whole mitochondrial genomes
You will have produced two trees for Bovidae (two different models), submit them as part of your practical as Figure 1 (5 marks/tree).
All text must be fully legible in your submitted trees to receive full marks.
Produce a Bayesean tree for the whole mitochondrial genomes of the Australidelphia order of marsupials (including all Australasian species and the South American Monito del Monte.
Multi-loci time-tree from Eldridge et al. 2018)
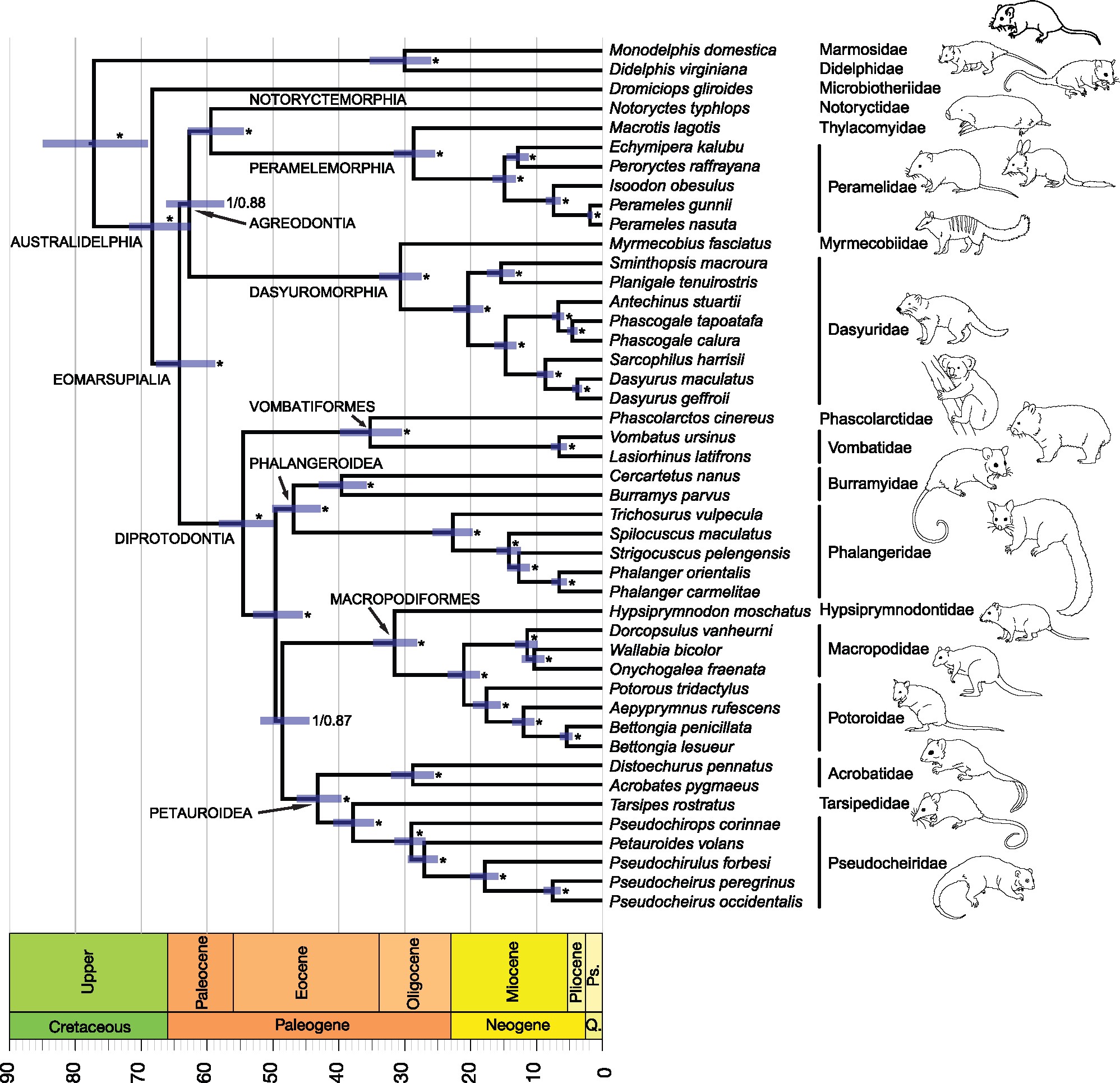
Submit the Australidelphia tree as Figure 2 (5 marks).
You can get the Australidelphia mitochondrial genomes by installing the fetch program on your VM from this location https://university-of-adelaide-bx-masters.github.io/BIOTECH-7005-BIOINF-3000/Practicals/evolutionary_prac/fetch using wget. You will need to chmod -x it before you can use it. You can download it to your Practical_5 subdirectory and run it from there. Fetch is a program that allows you to search the NCBI databases and batch download the results of your search; this is much more convenient than using a browser to retrieve things one by one. In order to get the sequences from NCBI you will need to run the command:
./fetch -email <youremailaddress> -query "mitochondrion[All Fields] AND \"Metatheria\"[Organism] NOT \"Didelphimorphia\"[Organism] AND \"complete genome\"[All fields] AND \"RefSeq\"[All fields]" -out metatheria_mtDNA.fa
The server requires your email to fetch the sequences and the -out <outfile> is required unless you want to stream the sequences to stdout.
Alternatively, these genomes can be obtained the way a biologist would from here.
- Reset the page view from 20 per page to 50 per page.
- Under the
Summarylink, selectFASTA (text) this will open a tab with all of the sequences as text - Copy the text in the browser window and paste it into a new document in the
sourcepane. - Save the source pane file into your
Practical_5subdirectory asmetatheria_mtDNA.fa
Q10. Does this tree agree with the known taxonomic groupings? If not, propose possible explanations.(1 mark)
Q11. Which two leaves are the closest related/have the shortest branch?(1 mark)
Q12. What is the evolutionary distance between these two leaves?(1 mark)
Q13. Based on your tree, which of the following two species are more closely related? Provide evidence supporting your answer.(1 mark)
1. Brush-tailed phascogale
2. Julia Creek dunnart
3. Yellow-footed antechinus
4. Numbat
Q14. Why is it that we chose to use the mitochondria to generate this tree? Comparing the topology of your trees to those in the provided literature, name a key factor that would affect the quality of a phylogeny made using a whole mitochondrial alignment. (2 marks)