BIOINF-3010-7150
Introduction to BLAST practical: Part 1 - Dave Adelson
We should really run a full course just on BLAST, but here we just show you how to use it for some common scenarios.
- Introduction to BLAST practical: Part 1 - Dave Adelson
1. Background
The original BLAST tool published/released in 1990 is one of the highest cited papers in the literature. The current version has been reengineered and is called BLAST+. NCBI BLAST is a suite of programs that will find local alignments of query sequences to database entries. We will use the command line version of BLAST to identify matching nucleotide or protein coding regions in a human genomic sequence and in the SwissProt database. You can find out about all things BLAST for command line BLAST here or in the BLAST Book.
BLAST can align a variety of sequence types:
- nucleotide sequences to nucleotide sequences (BLASTN)
- protein sequences to protein sequences (BLASTP)
- translated nucleotide sequences to protein sequences (BLASTX)
- protein sequences to translated nucleotide sequences (TBLASTN)
- translated nucleotide sequences to translated nucleotide sequences (TBLASTX)
There are a number of parameters that can be varied to increase sensitivity or speed up a search at the expense of sensitivity. There also numerous parameters to modify BLAST behaviour and customise output.
2. The BLAST algorithm
BLAST algorithm steps:
-
Generate a word list from the query sequence (W letters) Smaller values of W give a more sensitive search as the expense of speed
-
A word size of 5 would find no seeds to start an alignment in these sequences.
-
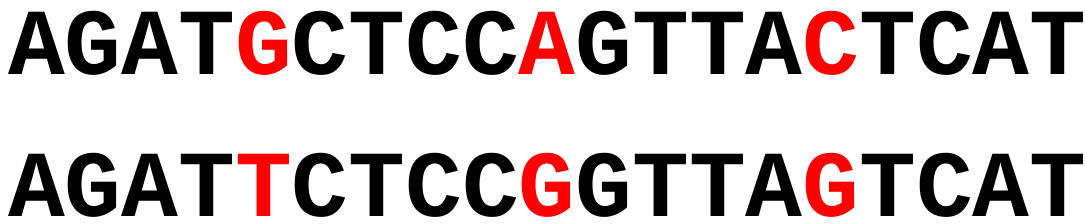
-
For each word, scan database for high scoring word matches (>T) T parameter only for protein alignments, smaller values of T give a more sensitive search at the expense of speed
-
Effect of T on word matches; X axis sequence 1 vs Y axis sequence 2 - Fig 5.3 BLAST Book
-
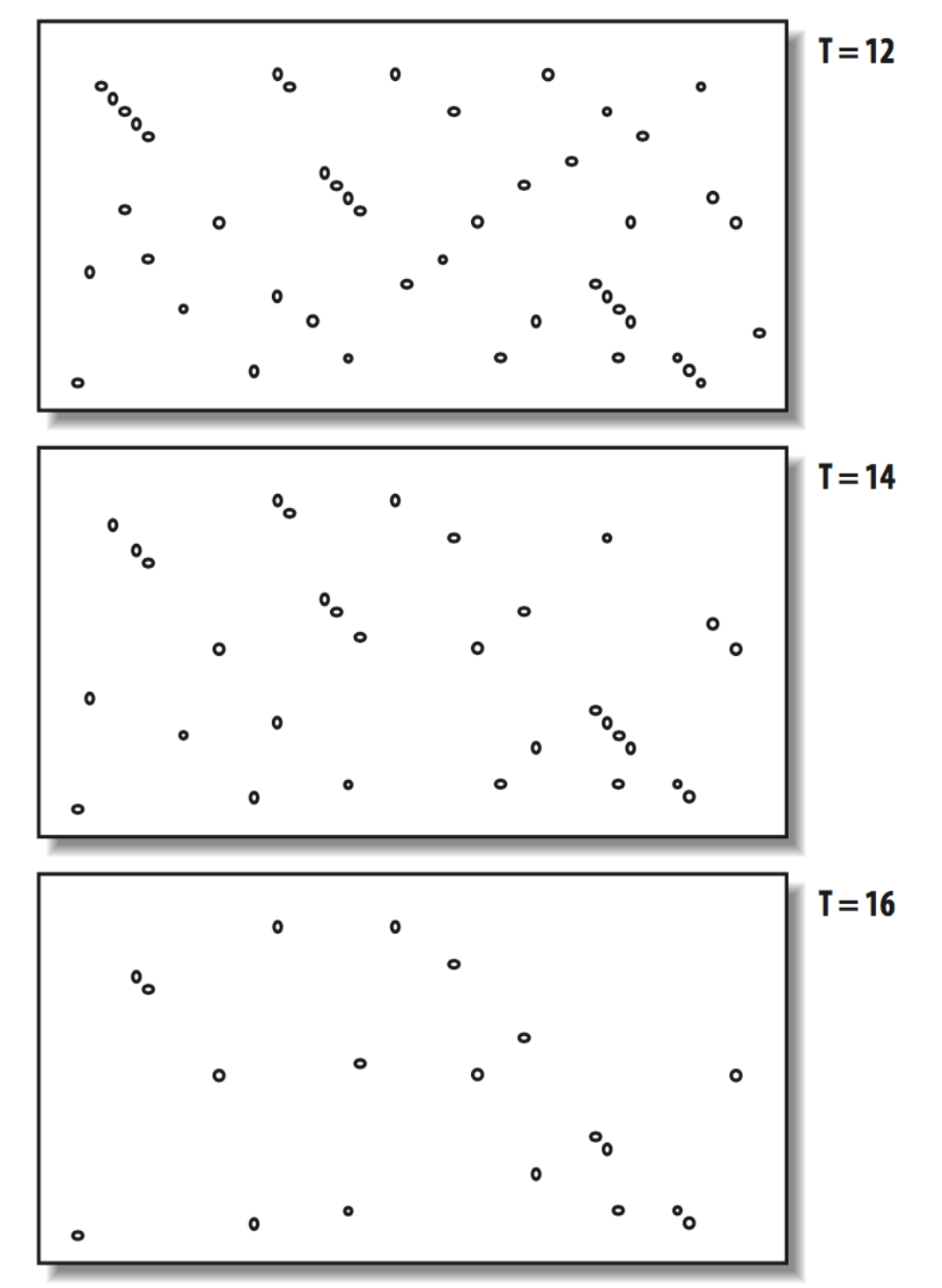
-
For each word, find high scoring neighbours and make word clusters
Word clusters - Fig 5.4 BLAST book
-

- Extend words/clusters to form High Scoring Pairs (HSP)
- Evaluate significance for each HSP
- Get local alignment for each HSP
- Report matches with low
e-value
3. Having a BLAST
This practical aims to familiarise you with the use of NCBI BLAST as a tool for annotation. You will use your VMs for this. Your first task will be to retrieve the databases you will require.
3.1 Activate NCBI BLAST environment
Open a terminal and at the bash command line prompt type the following to activate the conda environment for blast.
source activate bioinf
3.2 The data and project directory
The files you need are in ~/data/BLAST/.
See the README_BLAST.txt file for descriptions of the data files.
Make sure you create a working directory, along with useful sub-directories for the practical.
mkdir -p ~/Project_4/dbs
mkdir -p ~/Project_4/queries
mkdir -p ~/Project_4/results
Once you have created these directories copy the hg38.fa.gz, hg38_reduced.fa.gz, humanReps.fa.gz and uniprot_sprot.fasta.gz to ~/Project_4/dbs. Copy the remaining files to ~/Project_4/queries.
3.3 Prepare the BLAST databases
BLAST searches a special database of nucleotide sequences that have been broken into kmers/words for faster searching (it uses a hash table). It finds matching words in the database and then extends the matches to create a local alignment. You will need to format the BLAST databases that you will use.
You will need to decompress the hg 38.fa.gz, hg38_reduced.fa.gz and uniprot_sprot.fasta.gz files:
cd ~/Project_4/dbs
unpigz -p 2 uniprot_sprot.fasta.gz
unpigz -p 2 hg38.fa.gz
unpigz -p 2 hg38_reduced.fa.gz
- Can you think of a way to decompress these files using one command line instead of thre?
Once you have done this, you will need to index/format these files so that BLAST can search them.
The syntax for makeblastdb is as follows:
makeblastdb -in <reference.fa> -dbtype nucl -parse_seqids -out <database_name> -title "Database title"
In this example your input file is a nucleotide sequence file reference.fa so you need to specify the database type -dbtype nucl. You should use the -parse-seqids flag as it preserves the sequence identifiers in the input file. The -out flag specifies the name for db files and the -title flag is optional.
You will use the following command.
makeblastdb -in ~/Project_4/dbs/uniprot_sprot.fasta -dbtype 'prot' -parse_seqids -out ~/Project_4/dbs/sprot
This will generate six files that BLAST uses.
You will then need to index/format the human hg38 chromosome sequences so that BLAST can search them.
makeblastdb -in ~/Project_4/dbs/hg38.fa -dbtype 'nucl' -parse_seqids -out ~/Project_4/dbs/hg38
makeblastdb -in ~/Project_4/dbs/hg38_reduced.fa -dbtype 'nucl' -parse_seqids -out ~/Project_4/dbs/hg38_reduced
Now you are ready to familiarise yourself with command line BLAST. Once you make the blast dbs you can delete the original fasta files and recover some disk space
3.4 BLAST it!
For quick BLASTN help you can type:
blastn -help
For BLASTX:
blastx -help
Try these to see what the allowed syntax, flags and parameters are. You will probably want to pipe the output to less.
For detailed documentation for all things BLAST see here and for detailed command line options and flags see here
A typical command line might look something like this:
blastn -query [file.fasta] -task [megablast] -db [database file] -outfmt [0 through 17] -out [outputfile]
- For most analyses, I suggest using outfmt 7 and/or 17, 7 gives you a tab delimited file, 17 gives you a .sam file. However you should first familiarise yourself with the standard text output.
Output formatting options shown below. Feel free to experiment with these to see how they differ. Some of these are designed for compatibility with downstream applications that require specific input formats.
-outfmt
<number>
alignment view options:
0 = Pairwise,
1 = Query-anchored showing identities,
2 = Query-anchored no identities,
3 = Flat query-anchored showing identities,
4 = Flat query-anchored no identities,
5 = BLAST XML,
6 = Tabular,
7 = Tabular with comment lines,
8 = Seqalign (Text ASN.1),
9 = Seqalign (Binary ASN.1),
10 = Comma-separated values,
11 = BLAST archive (ASN.1),
12 = Seqalign (JSON),
13 = Multiple-file BLAST JSON,
14 = Multiple-file BLAST XML2,
15 = Single-file BLAST JSON,
16 = Single-file BLAST XML2,
17 = Sequence Alignment/Map (SAM),
18 = Organism Report
Note that when using output formats >4 some options for choosing the number and description of hits do not work or are incompatible (see below).
-num_descriptions
<Integer, >=0>
Number of database sequences to show one-line descriptions for
Not applicable for outfmt > 4
Default = `500’
* Incompatible with: max_target_seqs-num_alignments
<Integer, >=0>
Number of database sequences to show alignments for
Default = `250’
* Incompatible with: max_target_seqs-max_target_seqs
<Integer, >=1>
Maximum number of aligned sequences to keep
(value of 5 or more is recommended)
Default = `500’
* Incompatible with: num_descriptions, num_alignments
3.4.1 Alignments to Swissprot proteins
You can try the following
A “vanilla” BLAST command line that give the basic text output from BLAST, using
blastpfor a protein query to protein db.
blastp -query ~/Project_4/queries/Q967Q8.fasta -task blastp -db ~/Project_4/dbs/sprot
A command line that specifies a set of custom columns to report
blastp -query ~/Project_4/queries/Q967Q8.fasta -task blastp -db ~/Project_4/dbs/sprot -outfmt "7 delim= qaccver qlen sallgi sallacc slen pident length mismatch gapopen qstart qend sstart send evalue bitscore"
These will of course just dump everything to stdout but you know how to cope with that.
A command line that writes output to a file and specifies the number of threads to run and limits output to 50 targets. For a
blastxcommand that uses a nucleotide query against protein db.
blastx -query ~/Project_4/queries/hg38_gene_query.fasta -task blastx -db ~/Project_4/dbs/sprot -num_threads 2 -max_target_seqs 50 -out ~/Project_4/results/humgene_blastx_sprot.txt -outfmt 7
Call your output file whatever you like, as long as it makes sense to you.
Once BLASTX has completed you can look at your output using “head”, “less”, “more” or “cat” or open it with a text editor.
4. Effects of changing parameters
4.1 You can test the effect of -word_size on output and speed for blastn:
time blastn -query ~/Project_4/queries/hg38_gene_query.fasta -word_size 28 -db ~/Project_4/dbs/hg38_reduced -outfmt 7 -out ~/Project_4/results/W28.txt
time blastn -query ~/Project_4/queries/hg38_gene_query.fasta -word_size 11 -db ~/Project_4/dbs/hg38_reduced -outfmt 7 -out ~/Project_4/results/W11.txt
Be patient, this will take some time (around 8 minutes). While you are waiting, take some time to think about why this is taking so much longer.
4.2 You can test the effect of T -threshold (this is only for blastp) in conjunction with -word_size.
- First set T=21 and
-word_size= 2, we will reduce the wait time by using-num_threads=2 .
time blastp -query ~/Project_4/queries/multi-protein_query.fa -word_size 2 -threshold 21 -db ~/Project_4/dbs/sprot -num_threads 2 -outfmt 7 -max_target_seqs 1 -out ~/Project_4/results/W2T21multi.txt
- Now increase
-word_sizeto 7
time blastp -query ~/Project_4/queries/multi-protein_query.fa -word_size 7 -threshold 21 -db ~/Project_4/dbs/sprot -num_threads 2 -outfmt 7 -max_target_seqs 1 -out ~/Project_4/results/W7T21multi.txt
- keep
-word_size= 7 and lower T to 11
time blastp -query ~/Project_4/queries/multi-protein_query.fa -word_size 7 -threshold 11 -db ~/Project_4/dbs/sprot -num_threads 2 -outfmt 7 -max_target_seqs 1 -out ~/Project_4/results/W7T11multi.txt
- Now use a single protein query instead of multi-protein query
time blastp -query ~/Project_4/queries/Q967Q8.fasta -num_threads 2 -threshold 21 -db ~/Project_4/dbs/sprot -outfmt "7 delim= qaccver qlen sallgi sallacc slen pident length mismatch gapopen qstart qend sstart send evalue bitscore"
time blastp -query ~/Project_4/queries/Q967Q8.fasta -num_threads 2 -threshold 11 -db ~/Project_4/dbs/sprot -outfmt "7 delim= qaccver qlen sallgi sallacc slen pident length mismatch gapopen qstart qend sstart send evalue bitscore"
- What happened? Is parameter choice important? Why?
4.3 You can play with parameter combinations and with search types
Try other parameters for nucleotide comparisons, such as reward and penalty. These set the score or penalty for match vs mismatch during alignment. Default values for blastn are +2/-3. For increased sensitivity when searching less conserved sequences you can try +4/-5. Cost for gaps can be set with gapopen and gapextend and default values for blastn for these are 5 and 2, while default values for blastp/blastx are 11 and 1.
Try using different scoring matrices -matrix for protein alignments (BLASTP, BLASTX) such as blosum62 (default) vs blosum45 or blosum90.